2C-B
4-BROMO-2,5-DIMETOXIFENETYLAMIN
Syntes: En lösning av 100 g 2,5-dimetoxibensaldehyd i 220 g nitrometan behandlades med 10 g vattenfri ammoniumacetat och upphettades på ett ångbad i 2,5 timmar under tillfällig virvling. Den djupröda reaktionsblandningen strippades från överskottet av nitrometan under vakuum och återstoden kristalliserades spontant. Denna råa nitrostyren renades genom malning under IPA, filtrering och lufttorkning, vilket gav 85 g 2,5-dimetoxi-beta-nitrostyren som en gulorange produkt med tillräcklig renhet för nästa steg. Ytterligare rening kan åstadkommas genom omkristallisation från kokande IPA.
I en rundbottnad 2 L-kolv utrustad med en magnetomrörare och placerad under en inert atmosfär tillsattes 750 ml vattenfri THF, innehållande 30 g LAH. Därefter tillsattes, i THF-lösning, 60 g 2,5-dimetoxi-beta-nitrostyren. Den slutliga lösningen hade en smutsig gulbrun färg och hölls vid återflödestemperatur i 24 h. Efter kylning förstördes överskottet av hydrid genom droppvis tillsats av IPA. Därefter tillsattes 30 ml 15% NaOH för att omvandla de oorganiska fasta ämnena till en filtrerbar massa. Reaktionsblandningen filtrerades och filterkakan tvättades först med THF och sedan med MeOH. De kombinerade moderluten och tvättarna befriades från lösningsmedel under vakuum och återstoden suspenderades i 1,5 l H2O. Detta surgjordes med HCl, tvättades med 3x100 mL CH2Cl2, gjordes starkt basiskt med 25% NaOH och extraherades på nytt med 4x100 mL CH2Cl2. De sammanslagna extrakten strippades på lösningsmedel under vakuum, vilket gav 26 g oljig rest, som destillerades vid 120-130 °C vid 0,5 mm/Hg för att ge 21 g av en vit olja, 2,5-dimetoxifenetylamin (
2C-H) som mycket snabbt tar upp koldioxid från luften.
Till en väl omrörd lösning av 24,8 g 2,5-dimetoxifenetylamin i 40 mL isättika tillsattes 22 g elementärt brom löst i 40 mL ättiksyra. Efter ett par minuter bildades fasta ämnen och samtidigt utvecklades avsevärd värme. Reaktionsblandningen fick återgå till rumstemperatur, filtrerades och de fasta partiklarna tvättades sparsamt med kall ättiksyra. Detta var hydrobromidsaltet. Det finns många komplicerade saltformer, både polymorfer och hydrater, som kan göra isoleringen och karakteriseringen av 2C-B förrädisk. Den lyckligaste vägen är att bilda det olösliga hydrokloridsaltet med hjälp av den fria basen. Hela massan av ättiksyra-vått salt löstes i varmt H2O, gjordes basiskt till minst pH 11 med 25% NaOH och extraherades med 3x100 ml CH2Cl2. Avlägsnande av lösningsmedlet gav 33,7 g restprodukt som destillerades vid 115-130 °C vid 0,4 mm/Hg. Den vita oljan, 27,6 g, löstes i 50 mL H2O innehållande 7,0 g ättiksyra. Denna klara lösning omrördes kraftigt och behandlades med 20 mL koncentrerad HCl. Det vattenfria saltet av 2,5-dimetoxi-4-bromfenetylaminhydroklorid (2C-B) bildades omedelbart. Denna kristallmassa avlägsnades genom filtrering (den kan lossas avsevärt genom tillsats av ytterligare 60 mL H2O), tvättades med lite H2O och sedan med flera 50 mL portioner Et2O. Efter fullständig lufttorkning erhölls 31,05 g fina vita nålar, med ett mp på 237-239 °C med sönderdelning. När det finns för mycket H2O närvarande vid tillsatsen av den sista koncentrerade HCl erhålls en hydratiserad form av 2C-B. Hydrobromidsaltet smälter vid 214,5-215 °C. Acetatsaltet har rapporterats ha ett mp på 208-209 °C.
DOSERING: 12 - 24 mg.
Varaktighet
: 4-8 timmar.
DOM
STP; 2,5-DIMETOXI-4-METYLAMFETAMIN
SYNTEES: Till en lösning av 54,9 g 2,5-dimetoxi-4-metylbensaldehyd (se receptet för
2C-D för dess framställning) i 215 g isättika tillsattes 19,5 g vattenfri ammoniumacetat och 30,6 g nitroetan. Denna blandning upphettades under 3 timmar på ångbadet, reaktionsblandningen kyldes i ett vått isbad, vilket möjliggjorde spontan bildning av gula kristaller. Så mycket H2O som möjligt tillsattes (precis innan det blev en ihållande grumlig oljig karaktär) och efter ytterligare några timmars stående avlägsnades den kristallina 1-(2,5-dimetoxi-4-metylfenyl)-2-nitropropenen genom filtrering och omkristalliserades från kokande ättiksyra. Utbytet, efter torkning till konstant vikt, var 28,3 g och smp var 87-88 °C. Anal. (C12H15NO4) C, H ,N.
En suspension av 9,5 g LAH i 750 mL väl omrörd vattenfri Et2O hölls vid återflöde under en inert atmosfär, varvid returen av det kondenserade lösningsmedlet passerade genom en Soxhlet-blandare innehållande 9,5 g 1-(2,5-dimetoxi-4-metylfenyl)-2-nitropropen. Efter att tillsatsen av nitrostyren var fullständig hölls den omrörda suspensionen vid återflöde i ytterligare 4 timmar, kyldes sedan till rumstemperatur och fick fortsätta omrörningen över natten. Överskottet av hydrid förstördes genom tillsats av 750 ml 8% H2SO4, försiktigt tills väteutvecklingen upphörde, sedan med en hastighet som gjorde att de bildade fasta ämnena kunde dispergeras. Faserna separerades, vattenfasen tvättades en gång med Et2O, behandlades med 225 g kaliumnatriumtartrat och gjordes slutligen basisk (pH >9) med 5% NaOH. Detta extraherades med 3x150 mL CH2Cl2, extrakten poolades och lösningsmedlet avlägsnades under vakuum. Återstoden var 9,6 g av en klar olja som spontant bildade kristaller med ett mp på 60,5-61 °C från hexan. Dessa fasta ämnen löstes i 150 mL vattenfri Et2O och mättades med vattenfri HCl-gas. Efter att ha stått i rumstemperatur i 2 timmar avlägsnades den kristallina 2,5-dimetoxi-4-metylamfetaminhydrokloriden (DOM) genom filtrering, tvättades med Et2O och lufttorkades till konstant vikt. Det erhölls 8,25 g glittrande vita kristaller som hade ett mp på 190,5-191,5 °C. Sulfatet hade ett smp på 131 °C. Anal. (C12H20ClNO2) C, H ,N.
Ovanstående nitrostyren kan också omvandlas till den slutliga aminprodukten genom att motsvarande fenylaceton används som mellanprodukt. Till en väl omrörd suspension av 10,4 g järnpulver i 20 ml isättika som hölls vid återloppstemperatur tillsattes 4,9 g 1-(2,5-dimetoxi-4-metylfenyl)-2-nitropropen som ett fast ämne. Återflödet fortsatte i 2 timmar och sedan filtrerades allt genom våt Celite. Efter tvättning med 300 ml H2O följt av 300 ml Et2O separerades det kombinerade filtratet och tvättarna och den vattenhaltiga fasen extraherades med 2x100 ml Et2O. Den organiska fasen och extrakten kombinerades och tvättades med 2x100 mL mättat K2CO3 och lösningsmedlet avlägsnades under vakuum, vilket gav en rödaktig olja som vägde 3,3 g. Denna destillerades vid 111-115 °C vid 0,5 mm/Hg för att ge ett ljusgrönt fast ämne. Efter omkristallisering från bensen erhölls 2,8 g 1-(2,5-dimetoxi-4-metylfenyl)-2-propanon som vita kristaller med ett mp på 57-59 °C. Denna keton har också beskrivits som en blekgul olja med ett bp på 115-118 °C vid 0,4 mm/Hg. En lösning av 0,7 g 1-(2,5-dimetoxifenyl-4-metyl)-2-propanon i 20 mL MeOH behandlades med 6,0 g ammoniumacetat, 0,3 g natriumcyanoborhydrid och 3 g Linde 3 A molekylsikt. Blandningen omrördes över natten, de fasta partiklarna avlägsnades genom filtrering och filtratet löstes i 100 ml H2O. Lösningen surgjordes med utspädd H2SO4 och tvättades med 2x25 mL CH2Cl2. Vattenfasen gjordes basisk med vattenhaltig NaOH och produkten extraherades med 2x25 ml CH2Cl2. Lösningsmedlet avlägsnades under vakuum och återstoden destillerades (vid 160 °C vid 0,2 mm/Hg) för att ge en färglös produkt som löstes i 3 ml IPA, neutraliserades med koncentrerad HCl och späddes med 50 ml vattenfri Et2O. Man erhöll 0,18 g 2,5-dimetoxi-4-metylamfetaminhydroklorid (DOM) som ett vitt fast ämne med ett smp på 187-188 °C.
De optiska isomererna av DOM har framställts på två sätt. Den racemiska basen har upplösts som ortho-nitrotartranilsyrasalt genom omkristallisation från EtOH. (+)-syran ger företrädesvis (+)- eller "S"-isomeren av DOM. Den ovan nämnda 1-(2,5-dimetoxi-4-metylfenyl)-2-propanonen kan också amineras reduktivt med optiskt aktiv alfa-metylbensylamin med Raney Nickel. Denna amin isoleras och renas genom omkristallisation av hydrokloridsaltet. När den var optiskt ren avlägsnades bensylgruppen genom hydrogenolys med palladium på kol. Mp för någon av de optiska isomererna, som hydrokloridsalter, var 204-205 °C.
DOSERING: 3 - 10 mg.
Varaktighet
: 14 - 20 timmar.
MDA
3,4-METYLENDIOXIAMFETAMIN
SYNTETIS: (från piperonal) Till en lösning av 15,0 g piperonal i 80 ml isättika tillsattes 15 ml nitroetan följt av 10 g cyklohexylamin. Blandningen hölls vid ångbadstemperatur i 6 timmar, späddes med 10 ml H2O, seedades med en kristall av produkten och kyldes över natten vid 10 °C. De ljusgula kristallerna avlägsnades genom filtrering och lufttorkades för att ge 10,7 g 1-(3,4-metylendioxyfenyl)-2-nitropropen med ett mp på 93-94 °C. Detta höjdes till 97-98 °C genom omkristallisation från ättiksyra. Den mer konventionella nitrostyrensyntesen, där man använder ett överskott av nitroetan som lösningsmedel och vattenfri ammoniumacetat som bas, ger en oren produkt med mycket dåligt utbyte. Nitrostyren har framgångsrikt framställts från komponenterna i kall MeOH, med vattenhaltig NaOH som bas.
En suspension av 20 g LAH i 250 mL vattenfri THF placerades under inert atmosfär och omrördes magnetiskt. Droppvis tillsattes 18 g 1-(3,4-metylendioxyfenyl)-2-nitropropen i lösning i THF och reaktionsblandningen hölls vid återflöde i 36 h. Efter att ha återförts till rumstemperatur förstördes överskottet av hydrid med 15 mL IPA, följt av 15 mL 15% NaOH. Ytterligare 50 mL H2O tillsattes för att slutföra omvandlingen av aluminiumsalterna till ett löst, vitt, lättfiltrerat fast ämne. Detta avlägsnades genom filtrering och filterkakan tvättades med ytterligare THF. Det kombinerade filtratet och tvättarna strippades från lösningsmedel under vakuum och återstoden löstes i utspädd H2SO4. Tvättning med 3x75 mL CH2Cl2 avlägsnade mycket av färgen, och vattenfasen gjordes basisk och extraherades på nytt med 3x100 mL CH2Cl2. Avlägsnande av lösningsmedlet gav 13,0 g av en gulfärgad olja som destillerades. Fraktionen som kokade vid 80-90 °C vid 0,2 mm vägde 10,2 g och var vattenvit. Den löstes i 60 mL IPA, neutraliserades med koncentrerad HCl och späddes med 120 mL vattenfri Et2O vilket gav en bestående grumlighet. Kristaller bildades spontant som avlägsnades genom filtrering, tvättades med Et2O och lufttorkades för att ge 10,4 g 3,4-metylendioxiamfetaminhydroklorid (MDA) med ett mp på 187-188 °C.
(från 3,4-metylendioxifenylaceton) Till en lösning av 32,5 g vattenfri ammoniumacetat i 120 ml MeOH tillsattes 7,12 g 3,4-metylendioxifenylaceton (se under MDMA för dess framställning) följt av 2,0 g natriumcyanoborhydrid. Den resulterande gula lösningen omrördes kraftigt och koncentrerad HCl tillsattes regelbundet för att hålla pH i reaktionsblandningen mellan 6 och 7, vilket bestämdes med ett externt fuktigt universal-pH-papper. Efter flera dagar återstod oupplösta fasta ämnen i reaktionsblandningen och ingen mer syra behövdes. Reaktionsblandningen tillsattes till 600 mL utspädd HCl, och detta tvättades med 3x100 mL CH2Cl2. De kombinerade tvättarna extraherades tillbaka med en liten mängd utspädd HCl, de vattenhaltiga faserna kombinerades och gjordes basiska med 25% NaOH. Detta extraherades sedan med 3x100 mL CH2Cl2, dessa extrakt kombinerades och lösningsmedlet avlägsnades under vakuum för att ge 3,8 g av en rödfärgad rest. Denna destillerades vid 80-90 °C vid 0,2 mm/Hg för att ge 2,2 g av en absolut vattenvit olja. Det fanns ingen uppenbar bildning av ett karbonatsalt när den exponerades för luft. Detta löstes i 15 ml IPA, neutraliserades med 25 droppar koncentrerad HCl och späddes med 30 ml vattenfri Et2O. Långsamt utkristalliserades vita kristaller av 3,4-metylendioxiamfetaminhydroklorid (MDA) som vägde 2,2 g och hade ett mp på 187-188 °C. Framställningen av formamid (en prekursor till MDMA) och acetamid (en prekursor till MDE) beskrivs under dessa poster.
DOSERING: 80 - 160 mg.
VARAKTIGHET: 4 - 6 (
reviderad, september 2001).
MESKALIN;
3,4,5-TRIMETOXIFENETYLAMIN
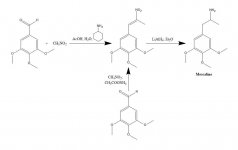
Syntes: En lösning av 20 g 3,4,5-trimetoxibensaldehyd, 40 mL nitrometan och 20 mL cyklohexylamin i 200 mL ättiksyra upphettades på ångbadet i 1 h. Reaktionsblandningen späddes sedan långsamt och under god omrörning med 400 mL H2O, vilket möjliggjorde bildandet av en tung gul kristallin massa. Denna avlägsnades genom filtrering, tvättades med H2O och sögs så torr som möjligt. Omkristallisering från kokande MeOH (15 mL/g) gav, efter filtrering och lufttorkning, beta-nitro-3,4,5-trimetoxistyren som ljusgula kristaller som vägde 18,5 g. En alternativ syntes var effektiv, med användning av ett överskott av nitrometan som lösningsmedel såväl som reagens, om mängden ammoniumacetatkatalys hölls liten. En lösning av 20 g 3,4,5-trimetoxibensaldehyd i 40 mL nitrometan innehållande 1 g vattenfri ammoniumacetat upphettades på ångbad i 4 h. Lösningsmedlet avlägsnades under vakuum och den kvarvarande gula oljan löstes i två volymer het MeOH, dekanterades från några olösliga ämnen och fick svalna. De kristaller som bildades avlägsnades genom filtrering, tvättades med MeOH och lufttorkades, vilket gav 14,2 g ljusgula kristaller av beta-nitro-3,4,5-trimetoxistyren. Användningen av dessa proportioner men med 3,5 g ammoniumacetat gav omfattande sidoreaktionsprodukter även när de bearbetades efter endast 1,5 timmars uppvärmning. Utbytet av nitrostyren var i detta senare fall otillfredsställande.
Till en svagt återflödande suspension av 2 g LAH i 200 ml Et2O tillsattes 2,4 g beta-nitro-3,4,5-trimetoxistyren som en mättad Et2O-lösning med hjälp av en Soxhlet extraktionskylare som modifierats för att tillåta kontinuerlig återföring av kondenserat lösningsmedel genom fingerborg. Efter att tillsatsen var fullständig bibehölls återflödesförhållandena i ytterligare 48 h. Efter kylning av reaktionsblandningen tillsattes försiktigt totalt 150 mL 1,5 N H2SO4, vilket förstörde överskottshydriden och slutligen gav två klara faser. Dessa separerades och den vattenhaltiga fasen tvättades en gång med 50 ml Et2O. Därefter tillsattes 50 g kaliumnatriumtartrat, följt av tillräckligt med NaOH för att pH skulle bli >9. Detta extraherades sedan med 3x75 mL CH2Cl2, och lösningsmedlet från de sammanslagna extrakten avlägsnades under vakuum. Återstoden destillerades vid 120-130 °C vid 0,3 mm/Hg, vilket gav en vit olja som löstes i 10 ml IPA och neutraliserades med koncentrerad HCl. De vita kristallerna som bildades späddes med 25 ml Et2O, avlägsnades genom filtrering och lufttorkades för att ge 2,1 g 3,4,5-trimetoxifenetylaminhydroklorid (M) som glittrande vita kristaller. Sulfatsaltet bildade spektakulära kristaller från vatten, men hade en bred och okarakteristisk mp. En alternativ syntes kan använda 3,4,5-trimetoxifenylacetonitril, enligt beskrivningen under beta-D.
DOSERING: 200-400 mg (som sulfatsalt), 178-356 mg (som hydrokloridsalt)
[Erowid Notera: Den ursprungliga texten läste "178-256" men detta var ett fel. Felet hittades av Bo och verifierades med Shulgin. Se Erowid Mescaline Dosage-sidan för en mer fullständig diskussion om formerna av meskalin].
VARAKTIGHET
: 10-12 h
TMA
3,4,5-TRIMETOXIAMFETAMIN
SYNTETIS: Till en lösning av 39,2 g 3,4,5-trimetoxibensaldehyd i 30 ml varm EtOH tillsattes 15,7 g nitroetan följt av 1,5 ml n-butylamin. Reaktionsblandningen fick stå vid 40 °C i 7 dagar. Efter kylning och skrapning erhölls fina gula nålar som, efter filtrering och lufttorkning, vägde 48 g. Omkristallisering från EtOH gav 2-nitro-1-(3,4,5-trimetoxifenyl)propen som gula kristaller med ett mp på 94-95 °C. Anal. (C12H15NO5) C,H,N. Alternativt behandlades en lösning av 20 g av aldehyden i 75 ml nitroetan med 4 g vattenfri ammoniumacetat och upphettades på ångbadet tills en djupröd färg hade genererats. Avlägsnande av överskottet av lösningsmedel/reagens under vakuum gav en röd olja som löstes i en lika stor volym kokande MeOH. Vid kylning separerades gula kristaller av nitropropenet. Omkristallisering från MeOH gav, efter lufttorkning till konstant vikt, 13,0 g med samma mp.
Under en inert atmosfär fuktades 38 g LAH med 100 mL vattenfri Et2O och suspenderades sedan i 1 L torr THF. Detta fördes upp till ett svagt återflöde och därefter tillsattes långsamt en lösning av 43,7 g 2-nitro-1-(3,4,5-trimetoxifenyl)propen i 160 mL THF. Återflödet fortsatte i 36 timmar och därefter kyldes reaktionsblandningen med ett externt isbad. Överskottet av hydrid förstördes genom försiktig tillsats av 38 ml H2O, vilket följdes av 38 ml 15% NaOH och slutligen ytterligare 114 ml H2O. De oorganiska salterna, som borde ha slutat som en lös, granulär, lättfiltrerbar massa, såg snarare ut som bibliotekspasta, men de filtrerades ändå. Man försökte tvätta med THF, men det var inte effektivt. Det kombinerade filtratet och tvättarna strippades på lösningsmedel under vakuum, vilket gav 31,5 g av den råa basen som en bärnstensfärgad olja. Denna löstes i 140 mL IPA, neutraliserades med koncentrerad HCl (15 mL krävdes) och späddes med 650 mL vattenfri Et2O. Det fanns en initial oljefas som under fortsatt omrörning övergick till ljusrosa fasta ämnen. Dessa finmaldes under CH3CN för att ge 15,2 g 3,4,5-trimetoxiamfetaminhydroklorid (TMA) som vita kristaller som smälte vid 195-211 °C. Alla aluminiumsalter från överallt löstes i utspädd HCl och 1 kg kaliumnatriumtartrat tillsattes. Därefter tillsattes 25 % NaOH, vilket gjorde att pH kunde sänkas till >9 utan utfällning av basisk aluminiumoxid. Extraktion av denna fas med CH2Cl2 följdes av avlägsnande av lösningsmedlet och saltbildning enligt beskrivningen ovan, vilket möjliggjorde isolering av ytterligare 6,4 g TMA. Den produkt som framställts på detta sätt innehåller ca 10-15% 3,5-dimetoxi-4-hydroxi-amfetamin som en förorening. En lösning av 20 g av den TMA som framställts på detta sätt i 200 ml 5% NaOH extraherades med 2x200 ml CH2Cl2. De poolade extrakten tvättades med 4x100 mL 5% NaOH, och de vattenhaltiga tvättarna poolades med den ursprungliga basfasen. Den organiska fasen strippades från sin CH2Cl2 under vakuum för att ge en olja som löstes i 40 mL IPA, neutraliserades med koncentrerad HCl och späddes med 400 mL vattenfri Et2O. Det bildades omedelbart spektakulära vita kristaller av ren 3,4,5-trimetoxiamfetaminhydroklorid, som vägde 15,4 g och hade ett mp på 220-221 °C. Vattenfasen bringades till neutralitet, behandlades med 10 g kaliumdivätefosfat, bringades till pH 9,0 genom försiktig tillsats av NaOH och extraherades med 5x100 ml CH2Cl2. Indunstning av lösningsmedlet under vakuum gav en olja som spontant kristalliserades. Denna produkt, 3,5-dimetoxi-4-hydroxi-amfetamin, kunde renas ytterligare genom
sublimering vid 130 °C vid 0,2 mm/Hg. Det var ett vitt kristallint fast ämne som långsamt missfärgades i luften. I litteraturen beskrivs ett pikratsalt med ett mp på 225 °C från EtOH.
DOSERING: 100 - 250 mg.
Varaktighet
: 6 - 8 timmar.