
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,949
- Solutions
- 3
- Reaction score
- 3,318
- Points
- 113
- Deals
- 1
Introduction
L'acide lysergique, le fragment de base dérivé des alcaloïdes de l'ergot de seigle, a été synthétisé en quatorze étapes, en commençant par le 3-bêta-carboxyéthylindole. Le produit de départ a été converti en un intermédiaire, le 1-benzoyl-5-céto-1,2,2a-3,4,5-hexahydrobenz-[cd]-indole (3), qui contient trois des quatre anneaux présents dans l'acide lysergique. Cette cétone a été transformée en un composé tétracyclique, la 9-céto-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4.3-fe]-quinoléine (8), puis en acide lysergique (14). Cette synthèse n'est pas simple et nécessite beaucoup d'expérience en laboratoire et de connaissances en chimie. Deplus, elle comporte plusieurs manipulations de substances dangereuses, qui doivent être effectuées avec des mesures de sécurité strictes.
Point d'ébullition : 536,2±50,0 °C à 760 mm Hg ;
Point de fusion : 240 °C ;
Poids moléculaire : 268,31 g/mole ;
Densité : 1,4 ± 0,1 g/mL ;
Numéro CAS : 82-58-6.
Équipement et verrerie :
- Réacteur d'hydrogénation en acier 2-3 L ;
- Autoclave en acier de 500 ml ;
- Échelle de laboratoire (0,01 - 500 g) ;
- Ballons à fond rond de 100, 200, 500 mL, 5 et 10 L ;
- Compresseur d'hydrogène (H2) et origine ;
- Fiole de Buchner et entonnoir (grand) 5 L [le filtre de Schott peut être utilisé pour de petites quantités] ;
- Machine Rotovap (grande) ;
- Source de vide;
- Ampoules à décanter de 500 ml et 2 l ;
- Ballon d'azote ~50-70 L (1 bar) ;
- Bouchons de septum pour les flacons ;
- Bain d'eau glacée salée ;
- 5 L x2, 2 L x2 ; 1 L x2 ; 500 mL x2 ; 100 mL x3 ; 50 mL x2 Béchers ;
- Seringue en verre ou pipette Pasteur ;
- Agitateur magnétique ou agitateur supérieur ;
- Dispositif de distillation sous vide ;
- Condenseur à reflux;
- Support d'autoclave et pince pour fixer l'appareil;
- Thermomètre de laboratoire (-20 °C à 200 °C) avec adaptateur pour ballon ;
- Papier indicateur de pH ;
- Baguette de verre et spatule ;
- Ampoule de 250 watts.
Réactifs.
- Acide 3-indolepropionique (1), 94,6 g (0,5 mol) ;
- 9,5 L d'eau distillée (H2O) ;
- ~400 g d'hydroxyde de sodium (NaOH) ;
- 116 g de nickel de Raney (Ni) ;
- 1050 mL d'acide chlorhydrique (HCl) concentré ;
- 2 mL Acide sulfurique (H2SO4 conc.) ;
- 210 mL Solution aq d'hydroxyde de sodium (NaOH) 12N ;
- 180 mL Chlorure de benzoyle ;
- ~1.5 L de méthanol (MeOH) ;
- ~1.6 L d'éthanol (EtOH) ;
- 201.2 mL Chlorure de thionyle (SOCl2) ;
- 1950 mL Disulfure de carbone (CS2) ;
- 240 g Chlorure d'aluminium (AlCl3) ;
- 2,5 L de benzène ;
- 500 mL d'hydroxyde de sodium 2N (NaOH) ;
- ~3.2 L d'éther diéthylique (Et2O) ;
- 3.3 L d'acide acétique glacial (AcOH) ;
- 352 g (1,1 mol) de bromhydrate de pyridine ;
- 5 L Chloroforme (CHCl3) ;
- ~1000 g Sulfate de magnésium (MgSO4) ;
- 307 g (2,35 mol) Méthylaminoacétone éthylène cétal (5) ;
- 4,5 L Benzène ;
- ~500 g Charbon actif (C) ;
- ~1 L Acétone ;
- ~500 g Bicarbonate de sodium (NaHCO3) ;
- 80 mL Anhydride acétique froid (Ac2O) ;
- 1,5 g Borohydrure de sodium (NaBH4) ;
- 75 mL Dioxyde de soufre (SO2 liquide) ;
- 40 g Cyanure de sodium (NaCN en poudre) ;
- 300 mL Cyanure d'hydrogène (HCN liquide) ;
- 78 mL Solution aqueuse d'hydroxyde de potassium (KOH) à 1,5 % ;
- 8,5 g d'arséniate de sodium hydraté ;
- ~ 50 mL Xylène ;
- 100 mL Solution diluée d'hydroxyde d'ammonium (NH4OH) ;
- 16,9 g Méthoxyde de sodium (MeONa).
Procédure
1-Benzoyl-3-(bêta-carboxyéthyl)-2,3-dihydroindole (2)L'acide 3-indolepropionique (1), 94,6 g (0,5 mol), est dissous dans 600 ml d'eau contenant 20 g d'hydroxyde de sodium. La solution a été mélangée à environ 100 g de catalyseur au nickel de Raney et hydrogénée à température ambiante dans une bombe d'hydrogénation en acier de 2-3 L à une pression H2 de 3000-4000 psi (207-276 bar). La réduction était généralement complète en 20-30 h, après quoi le catalyseur était filtré et lavé avec un peu d'eau. De l'acide HCl concentré, 85 ml, a été ajouté au filtrat et la solution a été refroidie. Si la réduction était incomplète, l'acide indolepropionique n'ayant pas réagi se séparait à ce stade et était éliminé par filtration. Le filtrat a ensuite été benzoylé selon la procédure habituelle de Schotten-Baumann, en utilisant 210 ml d'hydroxyde de sodium 12N et 180 ml de chlorure de benzoyle. La solution a été maintenue alcaline pendant toute la durée de la benzoylation et la température a été maintenue en dessous de 40 °C par refroidissement. Lorsque le chlorure de benzoyle a complètement réagi, le mélange a été refroidi et acidifié avec 300 ml d'acide HCl concentré. Le produit brut a été filtré et lavé à l'eau, puis extrait avec 4 portions de 1 L d'eau chaude pour éliminer l'acide benzoïque. Le produit sirupeux chaud (2), après décantation de l'extrait aqueux, a été cristallisé à partir de quelques volumes de méthanol ; rendement 103 g (70 %), MP : 151-153 °C.
1-Benzoyl-5-céto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (3)
1-Benzoyl-3-(bêta-carboxyéthyl)-2,3-dihydroindole (2), 118 g (0,4 mol), a été mélangé à 200 ml de chlorure de thionyle pur. La solution a été laissée au repos pendant 30 minutes, après quoi elle a été chauffée doucement pendant 15 à 20 minutes sur le bain de vapeur. L'excès de chlorure de thionyle a été complètement évaporé sous 30 °C dans le vide, et le chlorure d'acide brut a été dissous dans 200 ml de disulfure de carbone. La solution de chlorure d'acide a ensuite été ajoutée en un mince filet à une suspension vigoureusement agitée de 240 g de chlorure d'aluminium dans 1750 mL de disulfure de carbone contenue dans un ballon de 5 L (dans la HOUSSE !!!). Un complexe s'est séparé et l'agitation est devenue difficile. Le mélange a été chauffé à reflux et agité pendant une heure pour compléter la réaction, après quoi il a été décomposé très soigneusement en ajoutant 500 g de glace, 250 mL d'acide HCl conc. et 500 mL d'eau. Pendant la décomposition, l'agitation a été maintenue et le refroidissement a été affecté par la distillation périodique du disulfure de carbone dans le vide, et le produit a été extrait avec 2 L de benzène. L'extrait a été lavé soigneusement avec 500 ml d'hydroxyde de sodium 2N en trois portions, puis avec de l'eau. Il a été séché sur sulfate de magnésium et évaporé sous vide jusqu'à un petit volume. L'addition lente de plusieurs volumes d'éther a provoqué la cristallisation de la cétone jaune (3 ). Elle a été filtrée et lavée à l'éther ; rendement 85,3 g (77 %), MP : 146-147 °C. Un échantillon a été recristallisé pour analyse à partir de l'éther-benzène.
1-Benzoyl-3-(bêta-carboxyéthyl)-2,3-dihydroindole (2), 118 g (0,4 mol), a été mélangé à 200 ml de chlorure de thionyle pur. La solution a été laissée au repos pendant 30 minutes, après quoi elle a été chauffée doucement pendant 15 à 20 minutes sur le bain de vapeur. L'excès de chlorure de thionyle a été complètement évaporé sous 30 °C dans le vide, et le chlorure d'acide brut a été dissous dans 200 ml de disulfure de carbone. La solution de chlorure d'acide a ensuite été ajoutée en un mince filet à une suspension vigoureusement agitée de 240 g de chlorure d'aluminium dans 1750 mL de disulfure de carbone contenue dans un ballon de 5 L (dans la HOUSSE !!!). Un complexe s'est séparé et l'agitation est devenue difficile. Le mélange a été chauffé à reflux et agité pendant une heure pour compléter la réaction, après quoi il a été décomposé très soigneusement en ajoutant 500 g de glace, 250 mL d'acide HCl conc. et 500 mL d'eau. Pendant la décomposition, l'agitation a été maintenue et le refroidissement a été affecté par la distillation périodique du disulfure de carbone dans le vide, et le produit a été extrait avec 2 L de benzène. L'extrait a été lavé soigneusement avec 500 ml d'hydroxyde de sodium 2N en trois portions, puis avec de l'eau. Il a été séché sur sulfate de magnésium et évaporé sous vide jusqu'à un petit volume. L'addition lente de plusieurs volumes d'éther a provoqué la cristallisation de la cétone jaune (3 ). Elle a été filtrée et lavée à l'éther ; rendement 85,3 g (77 %), MP : 146-147 °C. Un échantillon a été recristallisé pour analyse à partir de l'éther-benzène.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
1-Benzoyl-2,2a,3,4-tétrahydro-4-[méthyl-(2-méthyl-1,2-dioxolan-2-yl-méthyl)-amino]-benz-[cd]-indol-5-(1H)-one (6)
Une solution de 270 g (0,76 mol) de 1-benzoyl-4-bromo-5-céto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole(4 ) et de 307 g (2.35 mol) de méthylaminoacétone éthylène cétal (5 ) dans 4500 mL de benzène sec ont été chauffés à reflux sous azote pendant 21 h dans 10 L RBF avec condenseur à reflux. Le mélange a été refroidi et 151 g (93,5 %) d'hydrobromure de méthylaminoacétone éthylène cétal ont été filtrés, MP : 158-159 °C.
Le filtrat a été lavé plusieurs fois avec de l'eau glacée, après quoi il a été extrait avec 2,5 L d'acide HCl dilué froid contenant 150 mL de l'acide concentré. Les extraits acides ont été immédiatement ajoutés à un excès d'hydroxyde de sodium dilué froid. Le produit a été extrait avec 1 L de chloroforme, et la solution chloroformique a été séchée sur sulfate de magnésium, traitée au charbon et concentrée sous vide. Le cétal-cétone résiduel (6) a été cristallisé à partir d'acétone ; MP : et mélange MP : 135-136 °C, rendement 220 g (71 %).
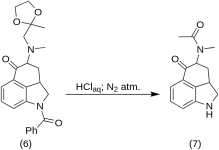
5-Céto-4-[N-méthyl-N-acétonylamino]-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7)
20 g de 1-benzoyl-2,2a,3,4-tétrahydro-4-[méthyl-(2-méthyl-1,3-dioxolan-2-yl-méthyl)-amino]-benz-[cd]-indol-5-(1H)-one (6 ) a été dissous dans un mélange de 250 ml d'acide HCl concentré et de 250 ml d'eau, et la solution a été maintenue sous azote à 37 °C dans 3-5 L RBF pendant cinq jours. Le mélange a été refroidi, traité au charbon, filtré et le filtrat a été concentré dans le vide jusqu'à obtention d'un petit volume. Le résidu a été traité avec un excès de bicarbonate de sodium ; le produit a été extrait avec du chloroforme froid et le solvant a été éliminé sous vide à température ambiante. La dicétone brute (7) a été réduite en poudre, mélangée à environ 75 ml de benzène-éther 1:1 et filtrée ; rendement : 9,8 g (77 %), MP : 105-107 °C. Unéchantillon pour analyse a été recristallisé à partir de l'éther benzénique ou de l'éthanol ; MP : 109-110 °C ; un monohydrochlorure a été obtenu à partir d'éthanol dilué ; MP : 200 °C déc.
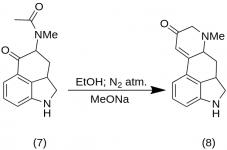
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (8)
25 g de 5-Keto-4-[N-methyl-N-acetonyl]-amino-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7 ) ont été mélangés avec 550 mL d'éthanol absolu. Le mélange a été agité sous azote et refroidi à -15 °C dans 2-5 L de RBF. Du méthoxyde de sodium, 16,9 g, a ensuite été ajouté et le mélange a été agité entre -10 °C et -12 °C pendant dix minutes. Le mélange réactionnel a été refroidi à -25 °C et le produit a été filtré sur un entonnoir de Buchner de 6,5 pouces et lavé avec un peu d'éthanol froid et d'éther. Avec une exposition minimale à l'air (contient du méthoxyde de sodium !), la cétone brute (8) a été immédiatement passée au slurry avec un peu d'eau glacée et filtrée à nouveau. Elle a été lavée avec de l'eau glacée, de l'éthanol et de l'éther ; rendement 16,2 g (69 %), MP : 145-147 °C. Un échantillon analytique a été recristallisé à partir d'éthanol dilué ; MP : 155-157 °C ; Le dihydrochlorure a été préparé et recristallisé à partir d'acétone aqueuse ; MP : 270 °C déc.
4-Acétyl-9-céto-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (9)
9-Céto-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (8), 24 g, ont été ajoutés à 80 mL d'anhydride acétique froid. Le mélange a été maintenu à 25 °C dans 200 mL de RBF pendant environ 5 minutes, après quoi il a été soigneusement refroidi, et le produit (9) a été filtré et lavé avec de l'éther ; rendement 20,5 g (76 %), mp : 167-170 °C. Une deuxième récolte a été obtenue par évaporation du filtrat, ce qui a porté le rendement total à 82 %. Un échantillon a été recristallisé à partir d'acétone-éthanol ; MP : 169-170 °C ; Le chlorhydrate a été préparé dans l'éthanol et a été recristallisé à partir d'éthanol aqueux ; MP : 250 °C déc.
9-Céto-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (8), 24 g, ont été ajoutés à 80 mL d'anhydride acétique froid. Le mélange a été maintenu à 25 °C dans 200 mL de RBF pendant environ 5 minutes, après quoi il a été soigneusement refroidi, et le produit (9) a été filtré et lavé avec de l'éther ; rendement 20,5 g (76 %), mp : 167-170 °C. Une deuxième récolte a été obtenue par évaporation du filtrat, ce qui a porté le rendement total à 82 %. Un échantillon a été recristallisé à partir d'acétone-éthanol ; MP : 169-170 °C ; Le chlorhydrate a été préparé dans l'éthanol et a été recristallisé à partir d'éthanol aqueux ; MP : 250 °C déc.
4-Acétyl-9-hydroxy-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (10)
10 g de 4-acétyl-9-céto-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroxindolo-[4,3-fg]-quinoléine (9 ) ont été ajoutés à un mélange de 150 mL de méthanol et 10 mL d'eau dans 500 mL de RBF. Du borohydrure de sodium, 1,5 g, a été ajouté, et la réaction a été laissée à température ambiante jusqu'à un petit volume, et un mélange de 15 ml d'acide HCl conc. et de 60 ml d'eau a été ajouté. Le chlorhydrate (10) qui s'est séparé en refroidissant a été filtré et lavé avec du méthanol, 9,0 g (79 %). Unéchantillon a été recristallisé à partir d'éthanol dilué ; MP : 245-246 °C dec.
Chlorhydrate de 4-acétyl-9-chloro-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (11)
Le chlorhydrate de 4-acétyl-9-hydroxy-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (10), 3.1 g, a été dissous dans 75 ml de dioxyde de soufre liquide contenu dans une gaine de verre dans un autoclave en acier de 500 ml. Le chlorure de thionyle, 1,2 ml, a été ajouté, et le récipient a été scellé et maintenu à 25 °C pendant 6 h. L'autoclave a été ventilé, et le mélange réactionnel a été retiré. Le dioxyde de soufre a été laissé s'évaporer tandis que le volume de la solution a été maintenu constant par l'ajout lent d'éther sec. Le chlorhydrate amorphe (11) a été filtré, lavé à l'éther et séché sous vide, MP : 130-135 °C dec. Rendement 3,5 g. L'utilisation de l'alcool 9-bêta-épimère dans cette réaction a donné le même chlorure avec un rendement comparable.
4-Acétyl-9-cyano-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (12)
Du cyanure de sodium sec et en poudre, 40 g, a été ajouté à 300 ml de cyanure d'hydrogène liquide glacé. Le mélange a été agité et refroidi dans la glace, et 7,5 g du chlorhydrate brut amorphe de 4-acétyl-9-chloro-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo [4,3f/g]-quinoléine (11 ) ci-dessus ont été ajoutés. L'agitation s'est poursuivie dans 500 ml de RBF pendant 30 minutes, après quoi le cyanure d'hydrogène a été rapidement distillé sous pression réduite à une température inférieure à 10 °C environ. Le résidu a été mélangé avec du chloroforme et de l'eau glacée, et le mélange résultant a été filtré. La couche organique a été séparée et la phase aqueuse a été extraite deux fois avec du chloroforme. Les extraits combinés ont été séchés sur sulfate de magnésium, décolorés et le solvant a été distillé dans le vide. Le produit (12) a été cristallisé à partir d'acétate d'éthyle ; rendement 3,3 g. (54% sur la base du chlorhydrate d'alcool), p.m. 172-174 °C. Larecristallisation à partir du même solvant a porté la p.m. à 181-182 °C.
Du cyanure de sodium sec et en poudre, 40 g, a été ajouté à 300 ml de cyanure d'hydrogène liquide glacé. Le mélange a été agité et refroidi dans la glace, et 7,5 g du chlorhydrate brut amorphe de 4-acétyl-9-chloro-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo [4,3f/g]-quinoléine (11 ) ci-dessus ont été ajoutés. L'agitation s'est poursuivie dans 500 ml de RBF pendant 30 minutes, après quoi le cyanure d'hydrogène a été rapidement distillé sous pression réduite à une température inférieure à 10 °C environ. Le résidu a été mélangé avec du chloroforme et de l'eau glacée, et le mélange résultant a été filtré. La couche organique a été séparée et la phase aqueuse a été extraite deux fois avec du chloroforme. Les extraits combinés ont été séchés sur sulfate de magnésium, décolorés et le solvant a été distillé dans le vide. Le produit (12) a été cristallisé à partir d'acétate d'éthyle ; rendement 3,3 g. (54% sur la base du chlorhydrate d'alcool), p.m. 172-174 °C. Larecristallisation à partir du même solvant a porté la p.m. à 181-182 °C.
9-Carbométhoxy-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (13)
Le produit (12 ) ci-dessus, 1,0 g, a été mélangé avec 15 mL de méthanol et 0,25 mL d'eau. Le mélange a été refroidi et 2 ml d'acide sulfurique concentré ont été ajoutés lentement. La solution a été scellée dans un tube de verre sous azote et chauffée à 100 °C pendant 23 à 24 h dans un RBF de 100 mL avec un condenseur à reflux. Le mélange a été traité avec du charbon décoloré et concentré sous vide jusqu'à environ 10 mL. Il a été versé sur un mélange de chloroforme (30 mL), de glace et de 10 g de bicarbonate de sodium. La couche de chloroforme a été séparée et la phase aqueuse a été extraite avec 3 portions de 10 mL de chloroforme. Les extraits combinés ont été séchés sur sulfate de magnésium, évaporés à sec et le produit (13) a été cristallisé à partir de benzène ; rendement 0,51 g (53 %), MP : 159-160 °C. Il a été recristallisé à partir d'acétate d'éthyle ; MP : 160-161 °C.
Le produit (12 ) ci-dessus, 1,0 g, a été mélangé avec 15 mL de méthanol et 0,25 mL d'eau. Le mélange a été refroidi et 2 ml d'acide sulfurique concentré ont été ajoutés lentement. La solution a été scellée dans un tube de verre sous azote et chauffée à 100 °C pendant 23 à 24 h dans un RBF de 100 mL avec un condenseur à reflux. Le mélange a été traité avec du charbon décoloré et concentré sous vide jusqu'à environ 10 mL. Il a été versé sur un mélange de chloroforme (30 mL), de glace et de 10 g de bicarbonate de sodium. La couche de chloroforme a été séparée et la phase aqueuse a été extraite avec 3 portions de 10 mL de chloroforme. Les extraits combinés ont été séchés sur sulfate de magnésium, évaporés à sec et le produit (13) a été cristallisé à partir de benzène ; rendement 0,51 g (53 %), MP : 159-160 °C. Il a été recristallisé à partir d'acétate d'éthyle ; MP : 160-161 °C.
Acide dl-lysergique synthétique (14)
Un mélange de 9-carbométhoxy-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (13), 3,9 g, et 78 ml d'une solution d'hydroxyde de potassium à 1,5 % a été porté à reflux pendant 30 minutes sous azote. De l'arséniate de sodium hydraté, 8,5 g, et du nickel de Raney (16 g, humide), préalablement désactivé par ébullition dans une suspension de xylène, ont été ajoutés, et le mélange a été chauffé à reflux et agité dans une atmosphère d'azote pendant 20 heures dans 200 mL de RBF avec un condenseur à reflux. La solution a été traitée au charbon et l'acide lysergique brut (14) a été précipité par neutralisation jusqu'à un pH de 5,6. Il a été filtré et lavé à l'eau ; rendement 1,04 g, MP : 240-242 °C dec. Une deuxième récolte, 0,16 g, MP : 233-235 °C dec. a également été obtenue ; rendement total 30%. L'acide a pu être purifié par dissolution dans l'hydroxyde d'ammonium dilué, traitement au charbon décolorant et reprécipitation au dioxyde de carbone, MP : 242-243 °C déc ; un mélange avec l'acide dl-lysergique fabriqué à partir de l'acide d-lysergique naturel a également donné 242-243 °C déc. L'acide anhydre a été obtenu par séchage sous vide pendant plusieurs heures à 150 °C.
Un mélange de 9-carbométhoxy-7-méthyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoléine (13), 3,9 g, et 78 ml d'une solution d'hydroxyde de potassium à 1,5 % a été porté à reflux pendant 30 minutes sous azote. De l'arséniate de sodium hydraté, 8,5 g, et du nickel de Raney (16 g, humide), préalablement désactivé par ébullition dans une suspension de xylène, ont été ajoutés, et le mélange a été chauffé à reflux et agité dans une atmosphère d'azote pendant 20 heures dans 200 mL de RBF avec un condenseur à reflux. La solution a été traitée au charbon et l'acide lysergique brut (14) a été précipité par neutralisation jusqu'à un pH de 5,6. Il a été filtré et lavé à l'eau ; rendement 1,04 g, MP : 240-242 °C dec. Une deuxième récolte, 0,16 g, MP : 233-235 °C dec. a également été obtenue ; rendement total 30%. L'acide a pu être purifié par dissolution dans l'hydroxyde d'ammonium dilué, traitement au charbon décolorant et reprécipitation au dioxyde de carbone, MP : 242-243 °C déc ; un mélange avec l'acide dl-lysergique fabriqué à partir de l'acide d-lysergique naturel a également donné 242-243 °C déc. L'acide anhydre a été obtenu par séchage sous vide pendant plusieurs heures à 150 °C.
Attachments
Last edited: