
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,796
- Solutions
- 3
- Reaction score
- 3,063
- Points
- 113
- Deals
- 1
Einführung
Lysergsäure, das von den Mutterkornalkaloiden abgeleitete Grundfragment, wurde in einer vierzehnstufigen Abfolge synthetisiert, beginnend mit 3-beta-Carboxyethylindol. Das Ausgangsmaterial wurde in das Zwischenprodukt 1-Benzoyl-5-keto-1,2,2a-3,4,5-hexahydrobenz-[cd]-indol (3) umgewandelt, das drei der vier in der Lysergsäure vorhandenen Ringe enthält. Dieses Keton wurde wiederum in die tetrazyklische Verbindung 9-Keto-7-Methyl-4,5,5a,6,6a,7,8,9-Octahydroindolo-[4.3-fe]-chinolin (8) und damit in Lysergsäure (14) umgewandelt. Diese Synthese ist nicht einfach und erfordert eine Menge Laborerfahrung und chemische Kenntnisse. Darüber hinaus gibt es mehrere Manipulationen mit gefährlichen Stoffen, die unter strengen Sicherheitsvorkehrungen durchgeführt werden müssen.
Siedepunkt: 536,2±50,0 °C bei 760 mm Hg;
Schmelzpunkt: 240 °C;
Molekulargewicht: 268,31 g/mol;
Dichte: 1,4 ± 0,1 g/ml;
CAS-Nummer: 82-58-6.
Ausrüstung und Glaswaren:
- Hydrierungsreaktor aus Stahl 2-3 L;
- Stahl-Autoklav 500 mL;
- Laborwaage (0,01 - 500 g ist geeignet);
- Rundbodenkolben 100, 200, 500 mL, 5 und 10 L;
- Wasserstoff (H2)-Kompressor und Quelle;
- Buchner-Kolben und Trichter (groß) 5 L [Schott-Filter kann für kleine Mengen verwendet werden];
- Rotovap-Maschine (groß);
- Vakuumquelle;
- Scheidetrichter 500 mL und 2 L;
- Stickstoffballon ~50-70 L (1 bar);
- Septumkappen für Kolben;
- Gesalzenes Eiswasserbad;
- 5 L x2, 2 L x2; 1 L x2; 500 mL x2; 100 mL x3; 50 mL x2 Bechergläser;
- Glasspritze oder Pasteurpipette;
- Magnetrührer oder Aufsatzrührer;
- Aufbau für die Vakuumdestillation;
- Rückflusskühler;
- Retortenständer und Klammer zur Befestigung der Apparatur;
- Laborthermometer (-20 °C bis 200 °C) mit Küvettenadapter;
- pH-Indikatorpapier;
- Glasstab und Spatel;
- 250-Watt-Glühbirne.
Reagenzien.
- 3-Indolpropionsäure (1), 94,6 g (0,5 mol);
- 9,5 L Destilliertes Wasser (H2O);
- ~400 g Natriumhydroxid (NaOH);
- 116 g Raney-Nickel (Ni);
- 1050 mL Chlorwasserstoffsäure (HCl) konzentriert;
- 2 mL Schwefelsäure (H2SO4 konz.);
- 210 mL 12N Natriumhydroxid (NaOH) wässrige Lösung;
- 180 mL Benzoylchlorid;
- ~1,5 L Methanol (MeOH);
- ~1,6 L Ethanol (EtOH);
- 201,2 mL Thionylchlorid (SOCl2);
- 1950 mL Schwefelkohlenstoff (CS2);
- 240 g Aluminiumchlorid (AlCl3);
- 2,5 L Benzol;
- 500 mL 2N Natriumhydroxid (NaOH);
- ~3,2 L Diethylether (Et2O);
- 3,3 L Eisessig (AcOH);
- 352 g (1,1 mol) Pyridinhydrobromid (Perbromid);
- 5 L Chloroform (CHCl3);
- ~1000 g Magnesiumsulfat (MgSO4);
- 307 g (2,35 mol) Methylaminoaceton-Ethylenketal (5);
- 4,5 L Benzol;
- ~500 g Aktivkohle (C);
- ~1 L Aceton;
- ~500 g Natriumbicarbonat (NaHCO3);
- 80 mL Kaltes Essigsäureanhydrid (Ac2O);
- 1,5 g Natriumborhydrid (NaBH4);
- 75 mL Schwefeldioxid (SO2 flüssig);
- 40 g Natriumcyanid (NaCN-Pulver);
- 300 mL Cyanwasserstoff (HCN flüssig);
- 78 mL 1,5%ige Kaliumhydroxid (KOH) wässrige Lösung;
- 8,5 g hydratisiertes Natriumarsenat;
- ~ 50 mL Xylol;
- 100 mL Ammoniumhydroxid (NH4OH) verdünnte Lösung;
- 16,9 g Natriummethoxid (MeONa).
Verfahren
1-Benzoyl-3-(beta-Carboxyethyl)-2,3-dihydroindol (2)3-Indolpropionsäure (1), 94,6 g (0,5 mol), wurde in 600 mL Wasser mit 20 g Natriumhydroxid gelöst. Die Lösung wurde mit etwa 100 g Raney-Nickel-Katalysator vermischt und bei RT in einer 2-3-L-Stahl-Hydrierbombe bei 3000-4000 psi (207-276 bar) H2-Druck hydriert. Die Reduktion war in der Regel nach 20-30 Stunden abgeschlossen, woraufhin der Katalysator filtriert und mit etwas Wasser gewaschen wurde. Dem Filtrat wurden 85 ml konzentrierte HCl-Säure zugesetzt, und die Lösung wurde abgekühlt. Wenn die Reduktion unvollständig war, trennte sich zu diesem Zeitpunkt nicht umgesetzte Indolpropionsäure ab und wurde durch Filtration entfernt. Das Filtrat wurde dann nach dem üblichen Schotten-Baumann-Verfahren mit 210 mL 12N Natriumhydroxid und 180 mL Benzoylchlorid benzoyliert. Die Lösung wurde während der gesamten Benzoylierung alkalisch gehalten, und die Temperatur wurde durch Kühlen unter 40 °C gehalten. Nach der vollständigen Umsetzung des Benzoylchlorids wurde das Gemisch abgekühlt und mit 300 mL konzentrierter HCl-Säure angesäuert. Das Rohprodukt wurde filtriert, mit Wasser gewaschen und anschließend mit 4 x 1 l heißem Wasser extrahiert, um die Benzoesäure zu entfernen. Das heiße, sirupartige Produkt (2) wurde nach Dekantieren des wässrigen Extrakts aus einigen Volumina Methanol kristallisiert; Ausbeute 103 g (70 %), MP: 151-153 °C.
1-Benzoyl-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indol (3)
1-Benzoyl-3-(beta-carboxyethyl)-2,3-dihydroindol (2), 118 g (0,4 mol), wurde mit 200 mL reinem Thionylchlorid versetzt. Die Lösung wurde 30 Min. stehen gelassen und dann 15-20 Min. auf dem Dampfbad leicht erwärmt. Überschüssiges Thionylchlorid wurde unter 30 °C im Vakuum vollständig eingedampft, und das rohe Säurechlorid wurde in 200 mL Schwefelkohlenstoff aufgelöst. Die Lösung des Säurechlorids wurde dann in einem dünnen Strahl zu einer kräftig gerührten Suspension von 240 g Aluminiumchlorid in 1750 mL Schwefelkohlenstoff in einem 5-Liter-Kolben (im HOOD!!!) gegeben. Es bildete sich ein Komplex, und das Rühren wurde schwierig. Das Gemisch wurde unter Rückfluss erhitzt und eine Stunde lang gerührt, um die Reaktion zu vervollständigen. Danach wurde es durch Zugabe von 500 g Eis, 250 mL konzentrierter HCl-Säure und 500 mL Wasser vorsichtig zersetzt. Während der Zersetzung wurde weiter gerührt, die Kühlung erfolgte durch periodische Destillation des Schwefelkohlenstoffs im Vakuum, und das Produkt wurde mit 2 l Benzol extrahiert. Der Extrakt wurde in drei Portionen mit 500 mL 2N-Natriumhydroxid und anschließend mit Wasser gewaschen. Er wurde über Magnesiumsulfat getrocknet und dann im Vakuum auf ein geringes Volumen eingedampft. Durch langsame Zugabe von mehreren Volumina Ether kristallisierte das gelbe Keton (3) aus. Es wurde filtriert und mit Ether gewaschen; Ertrag 85,3 g (77 %), MP: 146-147 °C. Eine Probe wurde zur Analyse aus Benzol-Ether umkristallisiert.
1-Benzoyl-3-(beta-carboxyethyl)-2,3-dihydroindol (2), 118 g (0,4 mol), wurde mit 200 mL reinem Thionylchlorid versetzt. Die Lösung wurde 30 Min. stehen gelassen und dann 15-20 Min. auf dem Dampfbad leicht erwärmt. Überschüssiges Thionylchlorid wurde unter 30 °C im Vakuum vollständig eingedampft, und das rohe Säurechlorid wurde in 200 mL Schwefelkohlenstoff aufgelöst. Die Lösung des Säurechlorids wurde dann in einem dünnen Strahl zu einer kräftig gerührten Suspension von 240 g Aluminiumchlorid in 1750 mL Schwefelkohlenstoff in einem 5-Liter-Kolben (im HOOD!!!) gegeben. Es bildete sich ein Komplex, und das Rühren wurde schwierig. Das Gemisch wurde unter Rückfluss erhitzt und eine Stunde lang gerührt, um die Reaktion zu vervollständigen. Danach wurde es durch Zugabe von 500 g Eis, 250 mL konzentrierter HCl-Säure und 500 mL Wasser vorsichtig zersetzt. Während der Zersetzung wurde weiter gerührt, die Kühlung erfolgte durch periodische Destillation des Schwefelkohlenstoffs im Vakuum, und das Produkt wurde mit 2 l Benzol extrahiert. Der Extrakt wurde in drei Portionen mit 500 mL 2N-Natriumhydroxid und anschließend mit Wasser gewaschen. Er wurde über Magnesiumsulfat getrocknet und dann im Vakuum auf ein geringes Volumen eingedampft. Durch langsame Zugabe von mehreren Volumina Ether kristallisierte das gelbe Keton (3) aus. Es wurde filtriert und mit Ether gewaschen; Ertrag 85,3 g (77 %), MP: 146-147 °C. Eine Probe wurde zur Analyse aus Benzol-Ether umkristallisiert.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
1-Benzoyl-2,2a,3,4-tetrahydro-4-[methyl-(2-methyl-1,2-dioxolan-2-yl-methyl)-amino]-benz-[cd]-indol-5-(1H)-on (6)
Eine Lösung von 270 g (0,76 mol) 1-Benzoyl-4-brom-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indol(4) und 307 g (2.35 mol) Methylaminoaceton-Ethylenketal (5) in 4500 mL trockenem Benzol wurden unter Stickstoff für 21 h in 10 L RBF mit Rückflusskühler refluxiert. Das Gemisch wurde abgekühlt und 151 g (93,5 %) Methylaminoaceton-Ethylenketal-Hydrobromid filtriert, MP: 158-159 °C.
Das Filtrat wurde mehrmals mit Eiswasser gewaschen und anschließend mit 2,5 L kalter, verdünnter HCl-Säure extrahiert, die 150 mL der konzentrierten Säure enthielt. Die Säureextrakte wurden sofort mit einem Überschuss an eiskaltem verdünntem Natriumhydroxid versetzt. Das Produkt wurde mit 1 l Chloroform extrahiert, und die Chloroformlösung wurde über Magnesiumsulfat getrocknet, mit Kohlenstoff behandelt und im Vakuum konzentriert. Das restliche Ketal-Keton (6) wurde aus Aceton kristallisiert; MP: und Gemisch MP: 135-136 °C, Ausbeute 220 g (71 %).
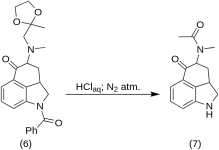
5-Keto-4-[N-methyl-N-acetonylamino]-1,2,2a,3,4,5-hexahydrobenz-[cd]-indol (7)
20 g 1-Benzoyl-2,2a,3,4-tetrahydro-4-[methyl-(2-methyl-1,3-Dioxolan-2-yl-methyl)-amino]-benz-[cd]-indol-5-(1H)-on (6) wurden in einem Gemisch aus 250 mL konzentrierter HCl-Säure und 250 mL Wasser gelöst, und die Lösung wurde fünf Tage lang unter Stickstoff bei 37 °C in 3-5 L RBF gehalten. Die Mischung wurde abgekühlt, mit Kohlenstoff behandelt, filtriert und das Filtrat im Vakuum auf ein kleines Volumen konzentriert. Der Rückstand wurde mit überschüssigem Natriumbicarbonat behandelt; das Produkt wurde mit kaltem Chloroform extrahiert, und das Lösungsmittel wurde bei RT im Vakuum entfernt. Das rohe Diketon (7) wurde pulverisiert, mit etwa 75 mL 1:1 Benzol-Ether aufgeschlämmt und filtriert; Ertrag 9,8 g (77 %), MP: 105-107 °C. Eine Probe zur Analyse wurde aus Benzol-Ether oder Ethanol umkristallisiert; MP: 109-110 °C; ein Monohydrochlorid wurde aus verdünntem Ethanol gewonnen; MP: 200 °C dez.
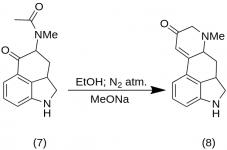
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (8)
25 g 5-Keto-4-[N-methyl-N-acetonyl]-amino-1,2,2a,3,4,5-hexahydrobenz-[cd]-indol (7) wurden mit 550 mL absolutem Ethanol versetzt. Die Mischung wurde unter Stickstoff gerührt und in 2-5 L RBF auf -15 °C abgekühlt. Dann wurde Natriummethoxid (16,9 g) zugegeben und das Gemisch zehn Minuten lang bei -10 °C bis -12 °C gerührt. Das Reaktionsgemisch wurde auf -25 °C abgekühlt, und das Produkt wurde über einen 6,5-Zoll-Buchtrichter filtriert und mit etwas kaltem Ethanol und Ether gewaschen. Das rohe Keton (8) wurde sofort mit etwas Eiswasser aufgeschlämmt und erneut filtriert, wobei man es so wenig wie möglich der Luft aussetzte (es enthält Natriummethoxid!). Es wurde mit Eiswasser, Ethanol und Ether gewaschen; Ertrag 16,2 g (69 %), MP: 145-147 °C. Eine analytische Probe wurde aus verdünntem Ethanol umkristallisiert; MP: 155-157 °C; Das Dihydrochlorid wurde hergestellt und aus wässrigem Aceton umkristallisiert; MP: 270 °C dez.
4-Acetyl-9-keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (9)
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (8), 24 g, wurde zu 80 mL kaltem Essigsäureanhydrid hinzugefügt. Die Mischung wurde bei 25 °C in 200 mL RBF für etwa 5 min gehalten, danach gründlich abgekühlt und das Produkt (9) filtriert und mit Ether gewaschen; Ausbeute 20,5 g (76 %), mp: 167-170 °C. Eine zweite Ernte wurde durch Eindampfen des Filtrats gewonnen; dadurch erhöhte sich die Gesamtausbeute auf 82 %. Eine Probe wurde aus Aceton-Ethanol umkristallisiert; MP: 169-170 °C; Das Hydrochlorid wurde in Ethanol hergestellt und aus wässrigem Ethanol umkristallisiert; MP: 250 °C dec.
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (8), 24 g, wurde zu 80 mL kaltem Essigsäureanhydrid hinzugefügt. Die Mischung wurde bei 25 °C in 200 mL RBF für etwa 5 min gehalten, danach gründlich abgekühlt und das Produkt (9) filtriert und mit Ether gewaschen; Ausbeute 20,5 g (76 %), mp: 167-170 °C. Eine zweite Ernte wurde durch Eindampfen des Filtrats gewonnen; dadurch erhöhte sich die Gesamtausbeute auf 82 %. Eine Probe wurde aus Aceton-Ethanol umkristallisiert; MP: 169-170 °C; Das Hydrochlorid wurde in Ethanol hergestellt und aus wässrigem Ethanol umkristallisiert; MP: 250 °C dec.
4-Acetyl-9-hydroxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (10)
10 g 4-Acetyl-9-keto-7-methyl-4,5,5a,6,6a,7,8,9-Octahydroxindolo-[4,3-fg]-chinolin (9) wurden zu einer Mischung aus 150 mL Methanol und 10 mL Wasser in 500 mL RBF gegeben. Natriumborhydrid (1,5 g) wurde zugegeben und die Reaktion bei RT bis zu einem kleinen Volumen ablaufen gelassen. Dann wurde ein Gemisch aus 15 mL konz. HCl-Säure und 60 mL Wasser zugegeben. Das Hydrochlorid (10) , das sich beim Abkühlen abtrennte, wurde filtriert und mit Methanol gewaschen, 9,0 g (79 %). Eine Probe wurde aus verdünntem Ethanol umkristallisiert; MP: 245-246 °C dez.
4-Acetyl-9-chlor-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolinhydrochlorid (11)
4-Acetyl-9-hydroxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolinhydrochlorid (10), 3.1 g, wurde in 75 mL flüssigem Schwefeldioxid in einem Glaseinsatz in einem 500 mL Stahlautoklaven gelöst. Thionylchlorid (1,2 ml) wurde hinzugefügt, das Gefäß verschlossen und 6 Stunden lang bei 25 °C gehalten. Der Autoklav wurde entlüftet und das Reaktionsgemisch entfernt. Man ließ das Schwefeldioxid verdampfen, während das Volumen der Lösung durch langsame Zugabe von trockenem Ether konstant gehalten wurde. Das amorphe Chlorhydrochlorid (11) wurde filtriert, mit Ether gewaschen und im Vakuum getrocknet, MP: 130-135 °C dez. Ausbeute 3,5 g. Die Verwendung des 9-beta-epimeren Alkohols in dieser Reaktion ergab das gleiche Chlorid in vergleichbarer Ausbeute.
4-Acetyl-9-cyano-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (12)
Trockenes, pulverisiertes Natriumcyanid, 40 g, wurde zu 300 mL eiskalter, flüssiger Blausäure gegeben. Das Gemisch wurde gerührt und in Eis gekühlt, und 7,5 g des rohen amorphen 4-Acetyl-9-chlor-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo [4,3f/g]-chinolinhydrochlorids (11) wurden zugegeben. In 500 mL RBF wurde 30 min lang gerührt, dann wurde die Cyanwasserstofflösung unter vermindertem Druck bei etwa 10 °C schnell abdestilliert. Der Rückstand wurde mit Chloroform und Eiswasser vermischt und das entstandene Gemisch filtriert. Die organische Schicht wurde abgetrennt, und die wässrige Phase wurde zweimal mit Chloroform extrahiert. Die kombinierten Extrakte wurden über Magnesiumsulfat getrocknet, entfärbt und das Lösungsmittel im Vakuum abdestilliert. Das Produkt (12) wurde aus Ethylacetat kristallisiert; Ausbeute 3,3 g (54 % insgesamt, bezogen auf das Alkoholhydrochlorid), m.p. 172-174 °C. DurchUmkristallisieren aus demselben Lösungsmittel wurde der m.p. auf 181-182 °C erhöht.
Trockenes, pulverisiertes Natriumcyanid, 40 g, wurde zu 300 mL eiskalter, flüssiger Blausäure gegeben. Das Gemisch wurde gerührt und in Eis gekühlt, und 7,5 g des rohen amorphen 4-Acetyl-9-chlor-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo [4,3f/g]-chinolinhydrochlorids (11) wurden zugegeben. In 500 mL RBF wurde 30 min lang gerührt, dann wurde die Cyanwasserstofflösung unter vermindertem Druck bei etwa 10 °C schnell abdestilliert. Der Rückstand wurde mit Chloroform und Eiswasser vermischt und das entstandene Gemisch filtriert. Die organische Schicht wurde abgetrennt, und die wässrige Phase wurde zweimal mit Chloroform extrahiert. Die kombinierten Extrakte wurden über Magnesiumsulfat getrocknet, entfärbt und das Lösungsmittel im Vakuum abdestilliert. Das Produkt (12) wurde aus Ethylacetat kristallisiert; Ausbeute 3,3 g (54 % insgesamt, bezogen auf das Alkoholhydrochlorid), m.p. 172-174 °C. DurchUmkristallisieren aus demselben Lösungsmittel wurde der m.p. auf 181-182 °C erhöht.
9-Carbomethoxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (13)
Das oben genannte Produkt (12) , 1,0 g, wurde mit 15 mL Methanol und 0,25 mL Wasser vermischt. Das Gemisch wurde abgekühlt und 2 mL konzentrierte Schwefelsäure langsam zugegeben. Die Lösung wurde in einem Glasröhrchen unter Stickstoff verschlossen und bei 100 °C für 23 bis 24 h in einem 100-mL-RBF mit Rückflusskühler erhitzt. Die Mischung wurde mit entfärbter Kohle behandelt und anschließend im Vakuum auf etwa 10 mL konzentriert. Es wurde auf eine Mischung aus Chloroform (30 mL), Eis und 10 g Natriumbicarbonat gegossen. Die Chloroformschicht wurde abgetrennt, und die wässrige Phase wurde mit 3 x 10 mL Chloroform extrahiert. Die vereinigten Extrakte wurden über Magnesiumsulfat getrocknet, zur Trockne eingedampft und das Produkt (13) wurde aus Benzol kristallisiert; Ausbeute 0,51 g (53 %), MP: 159-160 °C. Es wurde umkristallisiert aus Ethylacetat; MP: 160-161 °C.
Das oben genannte Produkt (12) , 1,0 g, wurde mit 15 mL Methanol und 0,25 mL Wasser vermischt. Das Gemisch wurde abgekühlt und 2 mL konzentrierte Schwefelsäure langsam zugegeben. Die Lösung wurde in einem Glasröhrchen unter Stickstoff verschlossen und bei 100 °C für 23 bis 24 h in einem 100-mL-RBF mit Rückflusskühler erhitzt. Die Mischung wurde mit entfärbter Kohle behandelt und anschließend im Vakuum auf etwa 10 mL konzentriert. Es wurde auf eine Mischung aus Chloroform (30 mL), Eis und 10 g Natriumbicarbonat gegossen. Die Chloroformschicht wurde abgetrennt, und die wässrige Phase wurde mit 3 x 10 mL Chloroform extrahiert. Die vereinigten Extrakte wurden über Magnesiumsulfat getrocknet, zur Trockne eingedampft und das Produkt (13) wurde aus Benzol kristallisiert; Ausbeute 0,51 g (53 %), MP: 159-160 °C. Es wurde umkristallisiert aus Ethylacetat; MP: 160-161 °C.
Synthetische dl-Lysergsäure (14)
Ein Gemisch aus 9-Carbomethoxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (13), 3,9 g, und 78 mL 1,5%iger Kaliumhydroxidlösung wurde 30 min lang unter Stickstoff refluxiert. Hydratisiertes Natriumarsenat, 8,5 g, und Raney-Nickel (16 g, feucht), das zuvor durch Kochen in Xylolsuspension deaktiviert worden war, wurden zugegeben, und das Gemisch wurde unter Rückfluss erhitzt und 20 Stunden lang in einer Stickstoffatmosphäre in 200 mL RBF mit Rückflusskühler gerührt. Die Lösung wurde mit Kohlenstoff behandelt, und die rohe Lysergsäure (14) wurde durch Neutralisation auf pH 5,6 ausgefällt. Sie wurde filtriert und mit Wasser gewaschen; Ausbeute 1,04 g, MP: 240-242 °C dez. Eine zweite Ernte, 0,16 g, MP: 233-235 °C dez. wurde ebenfalls erhalten; Gesamtausbeute 30%. DieSäure konnte durch Lösen in verdünntem Ammoniumhydroxid, Behandeln mit Entfärbungskohle und Umfällen mit Kohlendioxid gereinigt werden, MP: 242-243 °C dez; eine Mischung mit dl-Lysergsäure aus natürlicher d-Lysergsäure ergab ebenfalls 242-243 °C dez. Die wasserfreie Säure wurde durch mehrstündiges Trocknen im Vakuum bei 150 °C gewonnen.
Ein Gemisch aus 9-Carbomethoxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-chinolin (13), 3,9 g, und 78 mL 1,5%iger Kaliumhydroxidlösung wurde 30 min lang unter Stickstoff refluxiert. Hydratisiertes Natriumarsenat, 8,5 g, und Raney-Nickel (16 g, feucht), das zuvor durch Kochen in Xylolsuspension deaktiviert worden war, wurden zugegeben, und das Gemisch wurde unter Rückfluss erhitzt und 20 Stunden lang in einer Stickstoffatmosphäre in 200 mL RBF mit Rückflusskühler gerührt. Die Lösung wurde mit Kohlenstoff behandelt, und die rohe Lysergsäure (14) wurde durch Neutralisation auf pH 5,6 ausgefällt. Sie wurde filtriert und mit Wasser gewaschen; Ausbeute 1,04 g, MP: 240-242 °C dez. Eine zweite Ernte, 0,16 g, MP: 233-235 °C dez. wurde ebenfalls erhalten; Gesamtausbeute 30%. DieSäure konnte durch Lösen in verdünntem Ammoniumhydroxid, Behandeln mit Entfärbungskohle und Umfällen mit Kohlendioxid gereinigt werden, MP: 242-243 °C dez; eine Mischung mit dl-Lysergsäure aus natürlicher d-Lysergsäure ergab ebenfalls 242-243 °C dez. Die wasserfreie Säure wurde durch mehrstündiges Trocknen im Vakuum bei 150 °C gewonnen.
Attachments
Last edited:
