2C-B
4-BROM-2,5-DIMETHOXYPHENETHYLAMIN
SYNTHESE: Eine Lösung von 100 g 2,5-Dimethoxybenzaldehyd in 220 g Nitromethan wurde mit 10 g wasserfreiem Ammoniumacetat behandelt und 2,5 Stunden lang unter gelegentlichem Umschwenken auf einem Dampfbad erhitzt. Das tiefrote Reaktionsgemisch wurde unter Vakuum vom überschüssigen Nitromethan befreit, und der Rückstand kristallisierte spontan aus. Der Rückstand kristallisierte spontan aus. Dieses rohe Nitrostyrol wurde durch Zerkleinern unter IPA, Filtrieren und Lufttrocknen gereinigt, wobei 85 g 2,5-Dimethoxy-beta-nitrostyrol als gelb-oranges Produkt von ausreichender Reinheit für den nächsten Schritt erhalten wurden. Eine weitere Reinigung kann durch Umkristallisation aus siedendem IPA erreicht werden.
In einen mit einem Magnetrührer ausgestatteten 2-Liter-Rundkolben werden unter inerter Atmosphäre 750 mL wasserfreies THF, das 30 g LAH enthält, gegeben. Anschließend werden 60 g 2,5-Dimethoxy-beta-nitrostyrol in THF-Lösung zugegeben. Die fertige Lösung hatte eine schmutzig gelbbraune Farbe und wurde 24 Stunden lang bei Rückflusstemperatur gehalten. Nach dem Abkühlen wurde das überschüssige Hydrid durch tropfenweise Zugabe von IPA zerstört. Dann wurden 30 mL 15%ige NaOH zugegeben, um die anorganischen Feststoffe in eine filtrierbare Masse zu überführen. Das Reaktionsgemisch wurde filtriert und der Filterkuchen zunächst mit THF und dann mit MeOH gewaschen. Die vereinigten Mutterlaugen und Waschungen wurden unter Vakuum vom Lösungsmittel befreit und der Rückstand in 1,5 l H2O suspendiert. Dieses wurde mit HCl angesäuert, mit 3x100 mL CH2Cl2 gewaschen, mit 25% NaOH stark basisch gemacht und mit 4x100 mL CH2Cl2 reextrahiert. Die zusammengefassten Extrakte wurden im Vakuum vom Lösungsmittel befreit, was 26 g öligen Rückstand ergab, der bei 120-130 °C und 0,5 mm/Hg destilliert wurde und 21 g eines weißen Öls, 2,5-Dimethoxyphenethylamin
(2C-H), ergab, das sehr schnell Kohlendioxid aus der Luft aufnimmt.
Zu einer gut gerührten Lösung von 24,8 g 2,5-Dimethoxyphenethylamin in 40 mL Eisessig gab man 22 g elementares Brom, gelöst in 40 mL Essigsäure. Nach einigen Minuten kam es zur Bildung von Feststoffen und gleichzeitig zu einer erheblichen Wärmeentwicklung. Das Reaktionsgemisch wurde auf Raumtemperatur abgekühlt, filtriert und der Feststoff sparsam mit kalter Essigsäure gewaschen. Dies war das Hydrobromidsalz. Es gibt viele komplizierte Salzformen, sowohl Polymorphe als auch Hydrate, die die Isolierung und Charakterisierung von 2C-B tückisch machen können. Der einfachste Weg ist die Bildung des unlöslichen Hydrochloridsalzes über die freie Base. Die gesamte Masse des mit Essigsäure angefeuchteten Salzes wurde in warmem H2O gelöst, mit 25% NaOH auf mindestens pH 11 basisch gemacht und mit 3x100 mL CH2Cl2 extrahiert. Nach Entfernung des Lösungsmittels erhielt man 33,7 g Rückstand, der bei 115-130 °C und 0,4 mm/Hg destilliert wurde. Das weiße Öl, 27,6 g, wurde in 50 mL H2O mit 7,0 g Essigsäure gelöst. Diese klare Lösung wurde kräftig gerührt und mit 20 mL konzentrierter HCl behandelt. Es bildete sich sofort das wasserfreie Salz von 2,5-Dimethoxy-4-bromphenethylaminhydrochlorid (2C-B). Diese Kristallmasse wurde abfiltriert (sie kann durch Zugabe von weiteren 60 mL H2O erheblich gelockert werden), mit etwas H2O und anschließend mit mehreren 50 mL Et2O gewaschen. Nach vollständiger Lufttrocknung erhält man 31,05 g feiner weißer Nadeln mit einem mp-Wert von 237-239 °C unter Zersetzung. Wenn zum Zeitpunkt der Zugabe der letzten konzentrierten HCl zu viel H2O vorhanden ist, wird eine hydratisierte Form von 2C-B erhalten. Das Hydrobromidsalz schmilzt bei 214,5-215 °C. Für das Acetatsalz wurde eine Schmelztemperatur von 208-209 °C angegeben.
DOSIERUNG: 12 - 24 mg.
DAUER: 4 - 8 h.
DOM
STP; 2,5-DIMETHOXY-4-METHYLAMPHETAMIN
SYNTHESE: Zu einer Lösung von 54,9 g 2,5-Dimethoxy-4-methylbenzaldehyd (zur Herstellung siehe Rezeptur für
2C-D ) in 215 g Eisessig wurden 19,5 g wasserfreies Ammoniumacetat und 30,6 g Nitroethan gegeben. Dieses Gemisch wurde 3 Stunden lang auf dem Dampfbad erhitzt, dann wurde das Reaktionsgemisch in einem nassen Eisbad abgekühlt, so dass sich spontan gelbe Kristalle bildeten. Es wurde so viel H2O wie möglich zugegeben (kurz vor einem anhaltend trüben, öligen Charakter), und nach einigen weiteren Stunden Standzeit wurde das kristalline 1-(2,5-Dimethoxy-4-methylphenyl)-2-nitropropen durch Filtration abgetrennt und aus kochender Essigsäure umkristallisiert. Die Ausbeute nach dem Trocknen bis zur Gewichtskonstanz betrug 28,3 g und der mp-Wert 87-88 °C. Anal. (C12H15NO4) C, H ,N.
Eine Suspension von 9,5 g LAH in 750 mL gut gerührtem, wasserfreiem Et2O wurde unter inerter Atmosphäre am Rückfluss gehalten, wobei das kondensierte Lösungsmittel durch ein Soxhlet-Gefäß mit 9,5 g 1-(2,5-Dimethoxy-4-methylphenyl)-2-nitropropen zurückgeführt wurde. Nachdem die Zugabe des Nitrostyrols abgeschlossen war, wurde die gerührte Suspension weitere 4 Stunden am Rückfluss gehalten, dann auf Raumtemperatur abgekühlt und über Nacht weitergerührt. Das überschüssige Hydrid wurde durch Zugabe von 750 mL 8%iger H2SO4 zerstört, und zwar vorsichtig, bis die Wasserstoffentwicklung aufhörte, und dann mit einer Geschwindigkeit, die ein Dispergieren der gebildeten Feststoffe ermöglichte. Die Phasen wurden getrennt, die wässrige Phase einmal mit Et2O gewaschen, mit 225 g Kaliumnatriumtartrat behandelt und schließlich mit 5% NaOH basisch gemacht (pH >9). Es wurde mit 3x150 mL CH2Cl2 extrahiert, die Extrakte gepoolt und das Lösungsmittel unter Vakuum entfernt. Der Rückstand bestand aus 9,6 g eines klaren Öls, das spontan Kristalle mit einem mp von 60,5-61 °C aus Hexan bildete. Diese Feststoffe wurden in 150 mL wasserfreiem Et2O gelöst und mit wasserfreiem HCl-Gas gesättigt. Nach 2-stündigem Stehen bei Raumtemperatur wurde das kristalline 2,5-Dimethoxy-4-methylamphetaminhydrochlorid (DOM) durch Filtration abgetrennt, mit Et2O gewaschen und an der Luft bis zur Gewichtskonstanz getrocknet. Es wurden 8,25 g glitzernde weiße Kristalle mit einem mp-Wert von 190,5-191,5 °C erhalten. Das Sulfat hatte einen mp-Wert von 131 °C. Anal. (C12H20ClNO2) C, H ,N.
Das oben genannte Nitrostyrol kann auch über das entsprechende Phenylaceton in das endgültige Aminprodukt umgewandelt werden. Zu einer gut gerührten Suspension von 10,4 g pulverisiertem Eisen in 20 mL Eisessig, die bei Rückflusstemperatur gehalten wurde, wurden 4,9 g 1-(2,5-Dimethoxy-4-methylphenyl)-2-nitropropen als Feststoff hinzugefügt. Der Rückfluss wurde für 2 Stunden fortgesetzt und dann wurde alles durch nasses Celite filtriert. Nach dem Waschen mit 300 mL H2O und anschließend 300 mL Et2O wurden das kombinierte Filtrat und die Waschlösungen getrennt und die wässrige Phase mit 2x100 mL Et2O extrahiert. Die organische Phase und die Extrakte wurden vereinigt und mit 2x100 mL gesättigtem K2CO3 gewaschen, und das Lösungsmittel wurde unter Vakuum entfernt, wobei ein rötliches Öl mit einem Gewicht von 3,3 g erhalten wurde. Dieses wurde bei 111-115 °C und 0,5 mm/Hg destilliert, um einen blassgrünen Feststoff zu erhalten. Nach Umkristallisation aus Benzol erhielt man 2,8 g 1-(2,5-Dimethoxy-4-methylphenyl)-2-propanon als weiße Kristalle mit einem pH-Wert von 57-59 °C. Dieses Keton ist auch als blassgelbes Öl mit einem bp von 115-118 °C bei 0,4 mm/Hg beschrieben worden. Eine Lösung von 0,7 g 1-(2,5-Dimethoxyphenyl-4-methyl)-2-propanon in 20 mL MeOH wurde mit 6,0 g Ammoniumacetat, 0,3 g Natriumcyanoborhydrid und 3 g Linde 3 A Molekularsieben behandelt. Die Mischung wurde über Nacht gerührt, die Feststoffe durch Filtration entfernt und das Filtrat in 100 mL H2O gelöst. Die Lösung wurde mit verdünnter H2SO4 angesäuert und mit 2x25 mL CH2Cl2 gewaschen. Die wässrige Phase wurde mit wässriger NaOH basisch gemacht, und das Produkt wurde mit 2x25 mL CH2Cl2 extrahiert. Das Lösungsmittel wurde unter Vakuum entfernt und der Rückstand destilliert (bei 160 °C und 0,2 mm/Hg), um ein farbloses Produkt zu erhalten, das in 3 mL IPA aufgelöst, mit konzentrierter HCl neutralisiert und mit 50 mL wasserfreiem Et2O verdünnt wurde. Man erhielt 0,18 g 2,5-Dimethoxy-4-methylamphetaminhydrochlorid (DOM) als weißen Feststoff mit einer Temperatur von 187-188 °C.
Die optischen Isomere von DOM wurden auf zwei Arten hergestellt. Die racemische Base wurde durch Umkristallisation aus EtOH in das Salz der ortho-Nitrotartranilsäure aufgetrennt. Die (+)-Säure liefert vorzugsweise das (+)- oder "S"-Isomer von DOM. Auch das oben erwähnte 1-(2,5-Dimethoxy-4-methylphenyl)-2-propanon kann mit Raney-Nickel reduktiv mit optisch aktivem alpha-Methylbenzylamin aminiert werden. Dieses Amin wird isoliert und durch Umkristallisation des Hydrochloridsalzes gereinigt. Bei optischer Reinheit wurde die Benzylgruppe durch Hydrogenolyse mit Palladium auf Kohlenstoff entfernt. Der mp-Wert der beiden optischen Isomere in Form der Hydrochlorid-Salze lag bei 204-205 °C.
DOSIERUNG: 3 - 10 mg.
DAUER: 14 - 20 h.
MDA
3,4-METHYLENDIOXYAMPHETAMIN
SYNTHESE: (aus Piperonal) Zu einer Lösung von 15,0 g Piperonal in 80 mL Eisessig wurden 15 mL Nitroethan und 10 g Cyclohexylamin zugegeben. Das Gemisch wurde 6 h bei Dampfbadtemperatur gehalten, mit 10 mL H2O verdünnt, mit einem Produktkristall geimpft und über Nacht bei 10 °C abgekühlt. Die hellgelben Kristalle wurden abfiltriert und an der Luft getrocknet, um 10,7 g 1-(3,4-Methylendioxyphenyl)-2-nitropropen mit einer mp von 93-94 °C zu erhalten. Dieser Wert wurde durch Umkristallisation aus Essigsäure auf 97-98 °C erhöht. Die herkömmlichere Nitrostyrolsynthese mit einem Überschuss an Nitroethan als Lösungsmittel und wasserfreiem Ammoniumacetat als Base ergibt ein unreines Produkt in sehr geringer Ausbeute. Das Nitrostyrol wurde erfolgreich aus den Komponenten in kaltem MeOH mit wässrigem NaOH als Base hergestellt.
Eine Suspension von 20 g LAH in 250 mL wasserfreiem THF wird unter inerter Atmosphäre magnetisch gerührt. Man gab tropfenweise 18 g 1-(3,4-Methylendioxyphenyl)-2-nitropropen in Lösung in THF hinzu und hielt das Reaktionsgemisch 36 h lang am Rückfluss. Nachdem man es auf Raumtemperatur gebracht hatte, wurde das überschüssige Hydrid mit 15 mL IPA und anschließend mit 15 mL 15%iger NaOH vernichtet. Weitere 50 mL H2O wurden hinzugefügt, um die Umwandlung der Aluminiumsalze in einen lockeren, weißen, leicht filtrierbaren Feststoff abzuschließen. Dieser wurde durch Filtration entfernt, und der Filterkuchen wurde mit zusätzlichem THF gewaschen. Das kombinierte Filtrat und die Waschlösung wurden unter Vakuum abgetrennt und der Rückstand in verdünnter H2SO4 aufgelöst. Durch Waschen mit 3x75 mL CH2Cl2 wurde ein Großteil der Farbe entfernt, und die wässrige Phase wurde basisch gemacht und mit 3x100 mL CH2Cl2 reextrahiert. Nach Entfernung des Lösungsmittels erhielt man 13,0 g eines gelb gefärbten Öls, das destilliert wurde. Die Fraktion, die bei 80-90 °C bei 0,2 mm siedet, wog 10,2 g und war wasserweiß. Sie wurde in 60 mL IPA gelöst, mit konzentrierter HCl neutralisiert und mit 120 mL wasserfreiem Et2O verdünnt, was eine anhaltende Trübung ergab. Es bildeten sich spontan Kristalle, die durch Filtration abgetrennt, mit Et2O gewaschen und an der Luft getrocknet wurden, so dass man 10,4 g 3,4-Methylendioxyamphetaminhydrochlorid (MDA) mit einer mp von 187-188 °C erhielt.
(aus 3,4-Methylendioxyphenylaceton) Zu einer Lösung von 32,5 g wasserfreiem Ammoniumacetat in 120 mL MeOH wurden 7,12 g 3,4-Methylendioxyphenylaceton (Herstellung siehe unter MDMA) und 2,0 g Natriumcyanoborhydrid zugegeben. Die entstandene gelbe Lösung wurde kräftig gerührt, und es wurde regelmäßig konzentrierte HCl zugegeben, um den pH-Wert des Reaktionsgemischs zwischen 6 und 7 zu halten, wie mit einem externen feuchten Universal-pH-Papier bestimmt. Nach mehreren Tagen verblieben ungelöste Feststoffe in der Reaktionsmischung und es wurde keine weitere Säure benötigt. Das Reaktionsgemisch wurde mit 600 mL verdünnter HCl versetzt und mit 3x100 mL CH2Cl2 gewaschen. Die vereinigten Wässer wurden mit einer kleinen Menge verdünnter HCl rückextrahiert, die wässrigen Phasen vereinigt und mit 25 % NaOH basisch gemacht. Anschließend wurde mit 3x100 mL CH2Cl2 extrahiert, diese Extrakte vereinigt und das Lösungsmittel unter Vakuum entfernt, um 3,8 g eines rot gefärbten Rückstands zu erhalten. Dieser wurde bei 80-90 °C und 0,2 mm/Hg destilliert, um 2,2 g eines absolut wasserweißen Öls zu erhalten. An der Luft zeigte sich keine offensichtliche Bildung eines Carbonatsalzes. Dieses wurde in 15 mL IPA gelöst, mit 25 Tropfen konzentrierter HCl neutralisiert und mit 30 mL wasserfreiem Et2O verdünnt. Langsam bildeten sich weiße Kristalle von 3,4-Methylendioxyamphetaminhydrochlorid (MDA) mit einem Gewicht von 2,2 g und einer Temperatur von 187-188 °C. Die Herstellung von Formamid (einer Vorstufe von MDMA) und Acetamid (einer Vorstufe von MDE) ist unter diesen Einträgen beschrieben.
DOSIERUNG: 80 - 160 mg.
DAUER: 4 - 6
(überarbeitet, Sep 2001).
MESCALIN;
3,4,5-TRIMETHOXYPHENETHYLAMIN
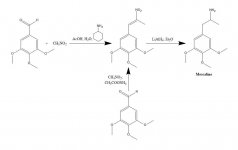
SYNTHESE: Eine Lösung von 20 g 3,4,5-Trimethoxybenzaldehyd, 40 mL Nitromethan und 20 mL Cyclohexylamin in 200 mL Essigsäure wurde 1 h lang auf dem Dampfbad erhitzt. Die Reaktionsmischung wurde dann langsam und unter gutem Rühren mit 400 mL H2O verdünnt, wodurch sich eine schwere gelbe kristalline Masse bildete. Diese wurde abfiltriert, mit H2O gewaschen und so trocken wie möglich abgesaugt. Die Umkristallisation aus siedendem MeOH (15 mL/g) ergab nach Filtration und Lufttrocknung beta-Nitro-3,4,5-Trimethoxystyrol als hellgelbe Kristalle mit einem Gewicht von 18,5 g. Eine alternative Synthese war effektiv, wobei ein Überschuss an Nitromethan als Lösungsmittel und Reagenz verwendet wurde, wenn die Menge an Ammoniumacetat-Katalysator gering gehalten wurde. Eine Lösung von 20 g 3,4,5-Trimethoxybenzaldehyd in 40 mL Nitromethan, die 1 g wasserfreies Ammoniumacetat enthielt, wurde 4 h lang auf dem Dampfbad erhitzt. Das Lösungsmittel wurde unter Vakuum abgestreift und das verbleibende gelbe Öl wurde in zwei Volumen heißem MeOH gelöst, von einigen Unlöslichkeiten dekantiert und abgekühlt. Die gebildeten Kristalle wurden abfiltriert, mit MeOH gewaschen und an der Luft getrocknet. Es entstanden 14,2 g hellgelbe Kristalle von beta-Nitro-3,4,5-trimethoxystyrol. Die Verwendung dieses Verhältnisses, jedoch mit 3,5 g Ammoniumacetat, ergab umfangreiche Nebenreaktionsprodukte, selbst wenn man nach nur 1,5 Stunden Erhitzen aufarbeitete. Die Ausbeute an Nitrostyrol war in diesem Fall nicht zufriedenstellend.
Zu einer unter leichtem Rückfluss stehenden Suspension von 2 g LAH in 200 mL Et2O wurden 2,4 g beta-Nitro-3,4,5-trimethoxystyrol in Form einer gesättigten Et2O-Lösung unter Verwendung eines Soxhlet-Extraktionskondensators zugegeben, der so modifiziert war, dass das kondensierte Lösungsmittel kontinuierlich durch den Fingerhut zurückgeführt werden konnte. Nach der vollständigen Zugabe wurden die Rückflussbedingungen für weitere 48 Stunden aufrechterhalten. Nach dem Abkühlen der Reaktionsmischung wurden vorsichtig insgesamt 150 mL 1,5 N H2SO4 zugegeben, wobei das überschüssige Hydrid zerstört wurde und schließlich zwei klare Phasen entstanden. Diese wurden getrennt, und die wässrige Phase wurde einmal mit 50 mL Et2O gewaschen. Dann wurden 50 g Kaliumnatriumtartrat zugegeben, gefolgt von ausreichend NaOH, um den pH-Wert auf >9 zu bringen. Anschließend wurde mit 3x75 mL CH2Cl2 extrahiert und das Lösungsmittel aus den zusammengefassten Extrakten unter Vakuum entfernt. Der Rückstand wurde bei 120-130 °C und 0,3 mm/Hg destilliert, wobei ein weißes Öl entstand, das in 10 mL IPA gelöst und mit konzentrierter HCl neutralisiert wurde. Die gebildeten weißen Kristalle wurden mit 25 mL Et2O verdünnt, abfiltriert und an der Luft getrocknet, um 2,1 g 3,4,5-Trimethoxyphenethylaminhydrochlorid (M) in Form glänzender weißer Kristalle zu erhalten. Das Sulfatsalz bildete spektakuläre Kristalle aus Wasser, hatte aber einen breiten und uncharakteristischen mp. Eine alternative Synthese kann mit 3,4,5-Trimethoxyphenylacetonitril durchgeführt werden, wie unter Beta-D beschrieben.
DOSIERUNG: 200-400 mg (als Sulfatsalz), 178-356 mg (als Hydrochloridsalz)
[Erowid-Anmerkung: Der ursprüngliche Text lautete "178-256", aber das war ein Fehler. Der Fehler wurde von Bo gefunden und mit Shulgin überprüft. Siehe die Erowid-Seite Meskalin-Dosierung für eine vollständigere Diskussion der Formen von Meskalin].
DAUER: 10-12 h
TMA
3,4,5-TRIMETHOXYAMPHETAMIN
SYNTHESE: Zu einer Lösung von 39,2 g 3,4,5-Trimethoxybenzaldehyd in 30 mL warmem EtOH wurden 15,7 g Nitroethan und 1,5 mL n-Butylamin zugegeben. Die Reaktionsmischung wurde 7 Tage lang bei 40 °C stehen gelassen. Durch Abkühlen und Kratzen wurden feine gelbe Nadeln erhalten, die nach Abfiltrieren und Lufttrocknen 48 g wogen. Durch Umkristallisieren aus EtOH erhielt man 2-Nitro-1-(3,4,5-trimethoxyphenyl)propen als gelbe Kristalle mit einer mp von 94-95 °C. Anal. (C12H15NO5) C,H,N. Alternativ wurde eine Lösung von 20 g des Aldehyds in 75 mL Nitroethan mit 4 g wasserfreiem Ammoniumacetat behandelt und auf dem Dampfbad erhitzt, bis sich eine tiefrote Farbe gebildet hatte. Das überschüssige Lösungsmittel/Reagenz wurde unter Vakuum entfernt, wobei ein rotes Öl entstand, das in einem gleichen Volumen siedendem MeOH gelöst wurde. Beim Abkühlen trennten sich gelbe Kristalle des Nitropropens ab. Die Umkristallisation aus MeOH ergab nach Trocknung an der Luft bis zur Gewichtskonstanz 13,0 g mit demselben mp.
Unter inerter Atmosphäre wurden 38 g LAH mit 100 mL wasserfreiem Et2O benetzt und dann in 1 L trockenem THF suspendiert. Dieses wurde unter leichtem Rückfluss zum Kochen gebracht und langsam eine Lösung von 43,7 g 2-Nitro-1-(3,4,5-trimethoxyphenyl)propen in 160 mL THF zugegeben. Der Rückfluss wurde 36 Stunden lang fortgesetzt, und dann wurde das Reaktionsgemisch in einem externen Eisbad abgekühlt. Das überschüssige Hydrid wurde durch vorsichtige Zugabe von 38 mL H2O zerstört, dann folgten 38 mL 15%ige NaOH und schließlich weitere 114 mL H2O. Die anorganischen Salze, die eigentlich eine lockere, körnige, leicht filtrierbare Masse hätten sein sollen, sahen eher wie eine Bibliothekspaste aus, wurden aber trotzdem filtriert. Es wurde versucht, mit THF zu waschen, was jedoch nicht gelang. Das kombinierte Filtrat und die Waschlösung wurden unter Vakuum vom Lösungsmittel befreit, wobei 31,5 g der rohen Base als bernsteinfarbenes Öl erhalten wurden. Dieses wurde in 140 mL IPA gelöst, mit konzentrierter HCl neutralisiert (15 mL wurden benötigt) und mit 650 mL wasserfreiem Et2O verdünnt. Es bildete sich eine ölige Anfangsphase, die sich unter weiterem Rühren in einen blassrosa Feststoff verwandelte. Diese wurden unter CH3CN fein gemahlen und ergaben 15,2 g 3,4,5-Trimethoxyamphetaminhydrochlorid (TMA) als weiße Kristalle, die bei 195-211 °C schmolzen. Alle Aluminiumsalze von überall wurden in verdünnter HCl gelöst, und 1 kg Kaliumnatriumtartrat wurde hinzugefügt. Durch Zugabe von 25 % NaOH konnte der pH-Wert auf >9 gebracht werden, ohne dass es zur Ausfällung von basischem Aluminiumoxid kam. Die Extraktion dieser Phase mit CH2Cl2 und die anschließende Entfernung des Lösungsmittels und die Salzbildung wie oben beschrieben ermöglichten die Isolierung von weiteren 6,4 g TMA. Das auf diese Weise hergestellte Produkt enthält etwa 10-15 % 3,5-Dimethoxy-4-hydroxyamphetamin als Verunreinigung. Eine Lösung von 20 g des auf diese Weise hergestellten TMA in 200 mL 5%iger NaOH wurde mit 2x200 mL CH2Cl2 extrahiert. Die zusammengefassten Extrakte wurden mit 4x100 mL 5%iger NaOH gewaschen, und die wässrigen Waschungen wurden mit der ursprünglichen Basisphase zusammengeführt. Die organische Phase wurde unter Vakuum von CH2Cl2 befreit, um ein Öl zu erhalten, das in 40 mL IPA aufgelöst, mit konzentrierter HCl neutralisiert und mit 400 mL wasserfreiem Et2O verdünnt wurde. Es bildeten sich sofort spektakuläre weiße Kristalle von reinem 3,4,5-Trimethoxyamphetaminhydrochlorid mit einem Gewicht von 15,4 g und einer Temperatur von 220-221 °C. Die wässrige Phase wurde zur Neutralität gebracht, mit 10 g Kaliumdihydrogenphosphat behandelt, durch vorsichtige Zugabe von NaOH auf pH 9,0 gebracht und mit 5x100 mL CH2Cl2 extrahiert. Die Verdampfung des Lösungsmittels im Vakuum ergab ein Öl, das spontan auskristallisierte. Dieses Produkt, 3,5-Dimethoxy-4-hydroxyamphetamin, konnte durch
Sublimation bei 130 °C und 0,2 mm/Hg weiter aufgereinigt werden. Es handelte sich um einen weißen kristallinen Feststoff, der sich an der Luft langsam verfärbte. In der Literatur wird ein Pikratsalz mit einer mp von 225 °C aus EtOH beschrieben.
DOSIERUNG: 100 - 250 mg.
DAUER: 6 - 8 h.